Using Samtools to index the bam file then visualize the alignment using IGV. Much of Galaxy-related features described in this section have been developed by.
Aligning Rna Seq Data Ngs Analysis
RNA-seq transcriptome sequencing is a very powerful method for transcriptomic studies that enables quantification of transcript levels as well as discovery of novel transcripts and transcript isoforms.

. RNA Analysis Tophat and set the parameters as follows. TopHat --library-type parameter HISAT2 --rna-strandness HTSeq --stranded-s Picard STRAND_SPECIFICITY option of CollectRnaSeqMetrics Kallisto quant StringTie. Find out the name of the computer that has been reserved for you httpscbsutccornelleduwwmachinesaspxi88.
This tutorial from 2017 covers the TopHat aligner. Align the RNA-seq short reads to a reference genome In the left tool panel menu under NGS Analysis select NGS. This should be correspond to every second file.
The requirements for aligning this type of data is slightly different from eg. There are several types of RNA-Seq. Using TophatCufflinksedgeR to analyze RNAseq data.
The newest version of Tophat can be invoked just as tophat in the command line without version string. Currently Ilumina sequencing produces longer reads for which the new version of Tophat should be used version 130. These lectures also cover UNIXLinux commands and some programming elements of R a popular freely available statistical software.
We recommend that you watch the video Aligning RNA-seq reads to reference genome instead which covers t. The Bowtie site provides pre-built indices for human mouse fruit fly and others. This tutorial is inspired by an exceptional RNA seq course at the Weill Cornell Medical College compiled by Friederike Dündar Luce Skrabanek and Paul Zumbo and by tutorials produced by Björn Grüning bgruening for Freiburg Galaxy instance.
If you would like to learn more about how to use vi try this tutorialgame. Type wq to save and quit vi. Everyone should have a BioHPC account to access the computer.
RNA Analysis Tophat and set the parameters as follows. TruSeq Strand Specific Total RNA. Go to an empty line with you cursor and copy paste the new RNA_HOME and PATH commands into the file.
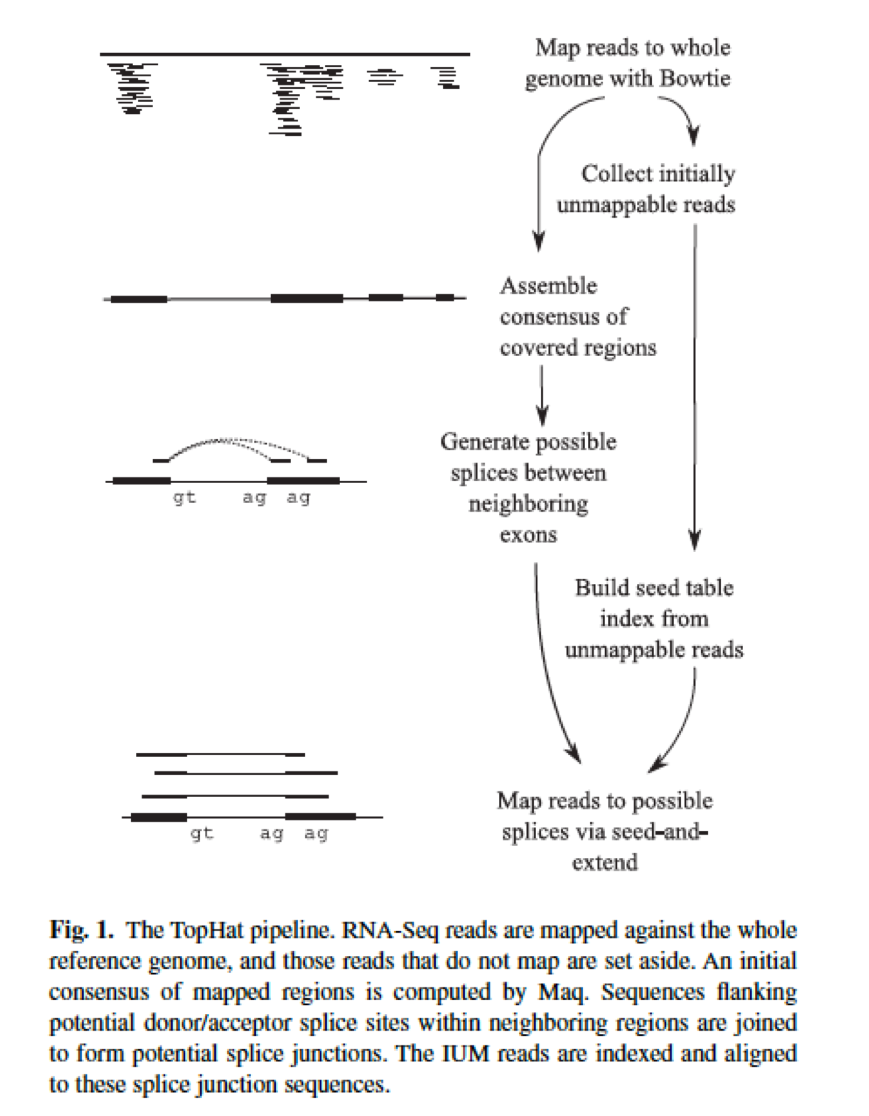
TopHat2 uses using Bowtie to align RNA-Seq reads to mammalian-sized genomes and then analyzes the mapping results to identify splice junctions between exons. Prepare the working directory. Previous version of Tophat 120.
Jeremy Goecks Galaxy RNAseq tutorial httpmaing2bxpsueduujeremypgalaxy-rna-seq-analysis-exercise. This is quite different conceptually to mapping to the transcriptome directly. Paired-end as individual datasets RNA-Seq FASTQ file forward reads.
A set of lectures in the Deep Sequencing Data Processing and Analysis module will cover the basic steps and popular pipelines to analyze RNA-seq and ChIP-seq data going from the raw data to gene lists to figures. Transcriptome splice-variantTSSUTR analysis microRNA-Seq etc. Is this single-end or paired-end data.
If you have Bowtie 2 installed and want to use it with Tophat v20 or later you must create Bowtie 2 indexes for your. RNA-seq Read Mapping with TOPHAT and STAR. Click on the multiple datasets icon and select all six of the FASTQ files.
It aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie included in this plugin and then analyzes the mapping results to identify splice junctions between exonsThis plugin runs on Mac OS and 64-bit Linux only it is not supported Windows. Is this single-end or paired-end data. In this tutorial well map reads from an RNA-seq study in Drosophila melanogaster to the reference genome using tophat.
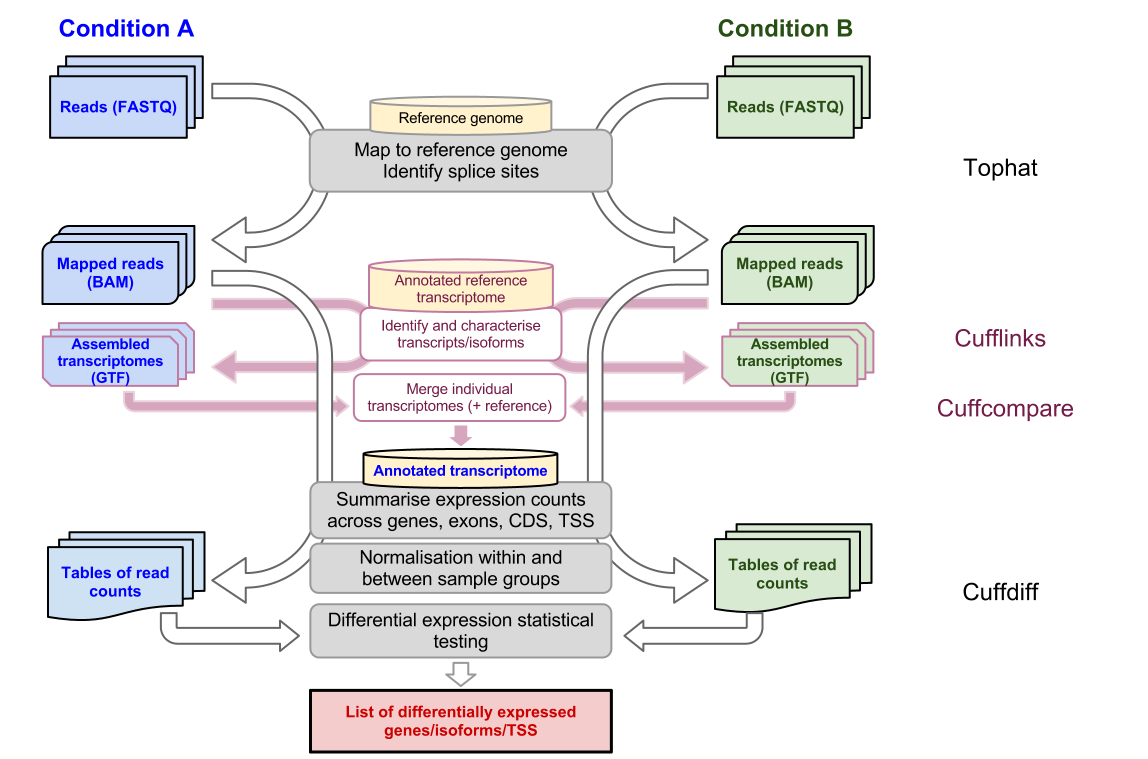
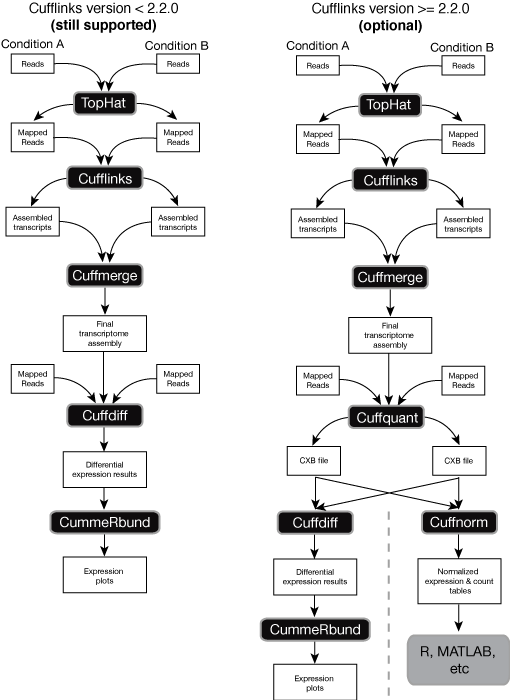
TopHat is designed to align RNA-seq reads to a reference genome while Cufflinks assembles these mapped reads into possible transcripts and then generates a final transcriptome assembly. To install TopHat from source package unpack the tarball and change directory to the package directory as follows. The guide below was adapted from a description of the method we initially developed for and applied in the RNA-Seq based Genome Annotation Assessment.
Configure the package specifying the install path and the library dependencies as needed. At the very end we can compare these results to the results we got from mapping directly to the. The user ID is normally your Cornell NetID.
RNA-Seq Tutorials Tutorial 1 RNA-Seq experiment design and analysis Instruction on individual software will be provided in other tutorials Tutorial 2 Advanced RNA-Seq Analysis topics Hands-on tutorials Analyzing human and potato RNA-Seq data using Tophat and Cufflinks in Galaxy. Cufflinks also includes Cuffdiff which accepts the reads assembled from two or more biological conditions and analyzes their differential expression of genes. Press the esc key to exit insert mode.
This practical will introduce some popular tools for basic processing of RNA-seq data. Some of the applications used in RNA sequencing analysis are the following. A very comprehensive tutorial of edgeR can be found in.
The real RNA-seq data would normally take several hours to process. In the left tool panel menu under NGS Analysis select NGS. Ad Build your Career in Data Science Web Development Marketing More.
This tutorial will focus on doing a 2 condition 1 replicate transcriptome analysis in mouse. This FASTQ files are RNA-seq data from two samples. If theres no index for your organism its easy to build one yourself.
Press the key to enter command mode. Flexible Online Learning at Your Own Pace. TopHat is a fast splice junction mapper for RNA-Seq reads.
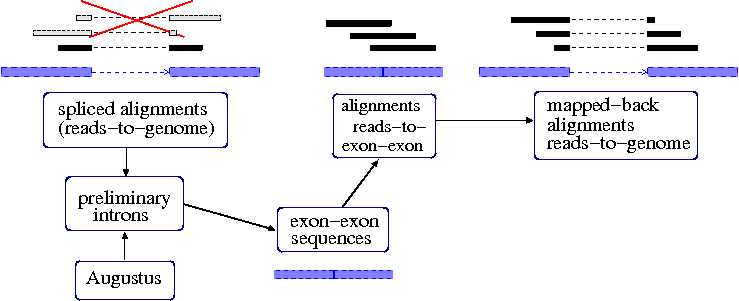
A_s_2_sequencetxtgz tophat_1log Step 4. Tophat Incorporating Illumina RNAseq into AUGUSTUS with TophatThis document describes a method for structurally annotating a genome based on deep sequencing of a transcriptome RNA-Seq. The following script creates the TopHat commands necessary for the alignments.
Map the reads to reference genome using TOPHAT. Background Web Resources. Special files were prepared for this workshop so that the exercise can be finished in minutes The files only include reads from first 20mb region from a genome.
RNA-Seq Tutorials Tutorial 1 RNA-Seq experiment design and analysis Instruction on individual software will be provided in other tutorials Tutorial 2 Hands-on using TopHat and Cufflinks in Galaxy Tutorial 3 Advanced RNA-Seq Analysis topics. To find junctions with TopHat youll first need to install a Bowtie index for the organism in your RNA-Seq experiment. Click on the multiple datasets icon and select all six of the forward FASTQ files ending in 1fastq.
Reference-Based RNA-Seq Data Analysis Workshop Session 2 Exercise. Bowtie1 is an ultrafast memory-effi cient aligner for large sets of short reads. Tophatcmdwithmetadatapastetophat -G gtf -p 5 -o outputdir libraryName bowidx fastqDirfastqnnnnsep sinkfiletophat-commandsshtypeoutput catbinsh nnnn cattophatcmd sink.
Press the i key to enter insert mode. Invest 2-3 Hours A Week Advance Your Career. Tophat is a splicing aware aligner so we can map transcripts to the genome.

Tophat Cufflinks Command Pipeline
Introduction To Bulk Rnaseq Analysis Bioinformatics Documentation

Rna Seq Alignment And Visualization Youtube

Reference Based Rnaseq Data Analysis Long
Bioinformatics Greifswald Incorporatingrnaseq Tophat
The Cufflinks Rna Seq Workflow

Basic Analyses With Tophat Cufflinks Rnaseq Tutorial 1 Documentation

Rna Seq Course Alignment Using Tophat Old Youtube
0 comments
Post a Comment